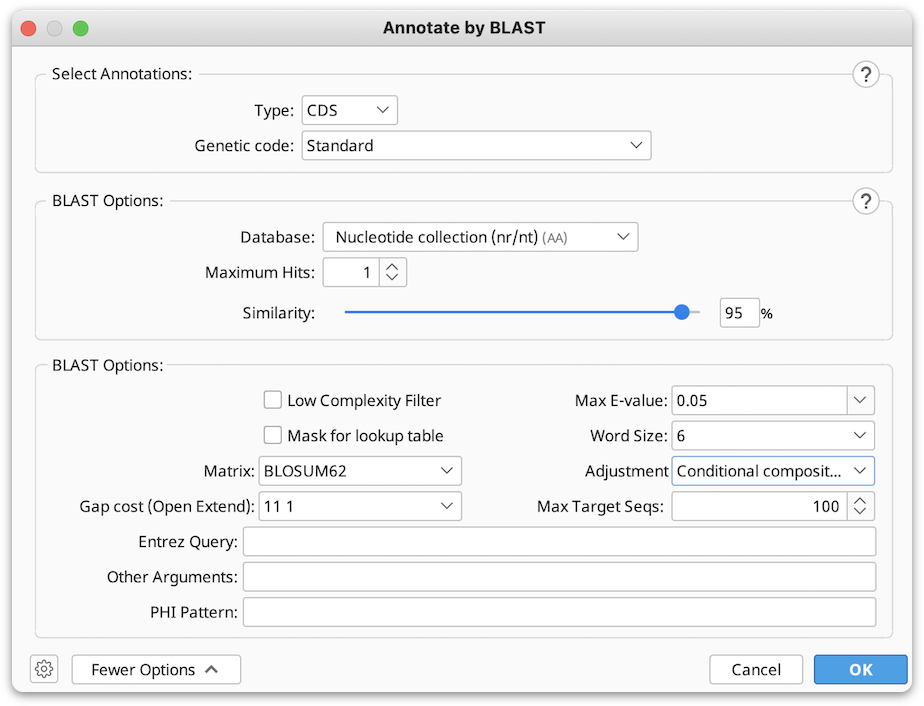
If you wish to use BLAST to annotate previously identified ORF, CDS or mRNA regions on your sequence, you can use the Annotate by BLAST tool.
This function will find and extract all the annotations of the selected type, translate them, and run blastp against a selected BLAST db. Annotations from the BLAST hits are then back translated and transferred directly onto your sequence if they meet the similarity threshold.
To annotate your sequences using Annotate by BLAST option :
1. Annotate your sequence(s) with ORF, CDS or mRNA annotations, either using :
- Find ORF option under Annotate and Predict or Glimmer plugin to add ORF annotations.
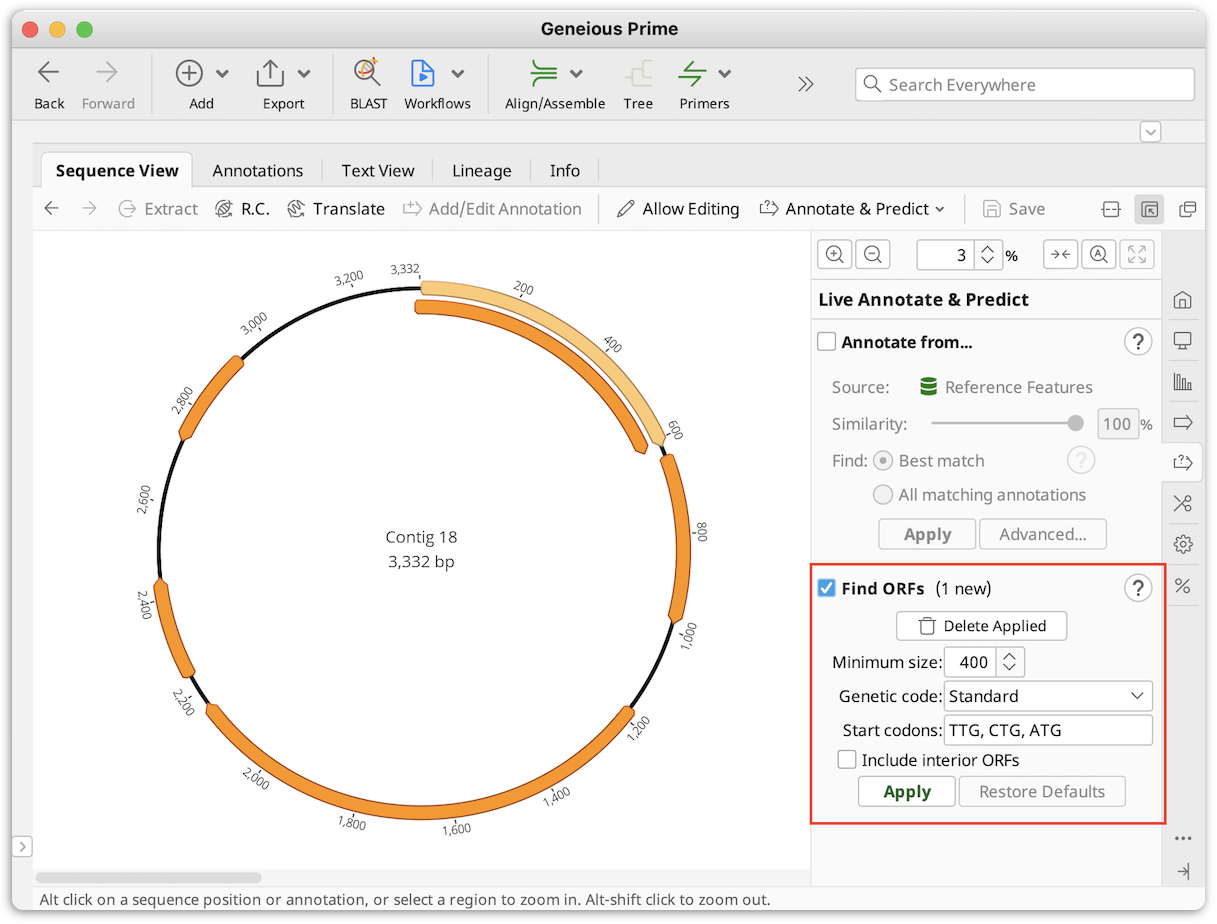
-
- Augustus (available as a plugin) for CDS or mRNA.

2. Select your annotated nucleotide sequence and choose Annotate by BLAST tool under the Annotate and Predict menu.
In the window that appears select the type of annotations on your sequence (ORF’s, CDS or mRNA) from the drop down menu, and set the Genetic code for your sequence.
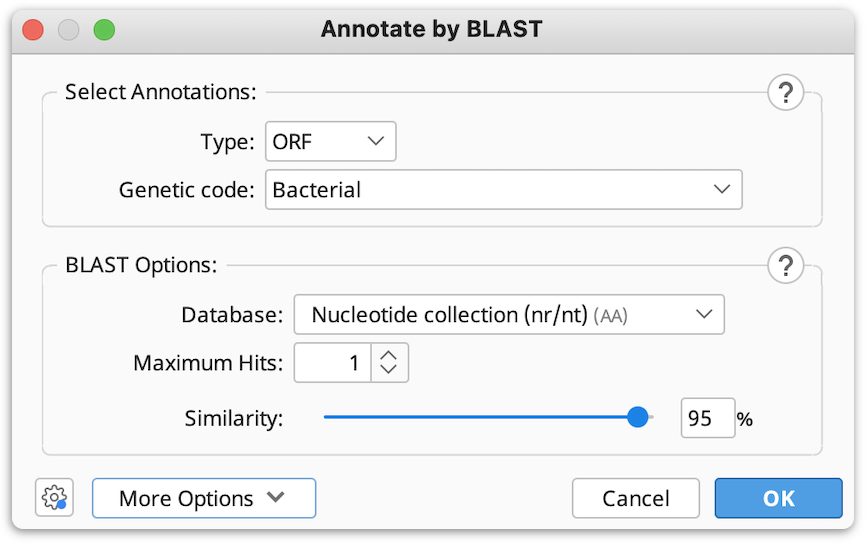
3. Select the BLAST database you wish to BLAST against (only amino acid databases can be selected, as BLASTp will be used to find the matches for your annotations).
You can choose the number of BLAST hits to return under Maximum Hits option (if more than 1 hit is selected and the hits cover the same region of sequence, only the top hit will be annotated).
4. If necessary, adjust the Similarity slider to set the threshold for finding matches between your translated annotations and the BLAST hits.
5. Click OK to start the BLAST search (you can continue to use Geneious while BLAST search is being performed).
Once the BLAST search is finished, annotations from the BLAST hits will be back-translated and transferred on to your sequence if they are above the similarity threshold.
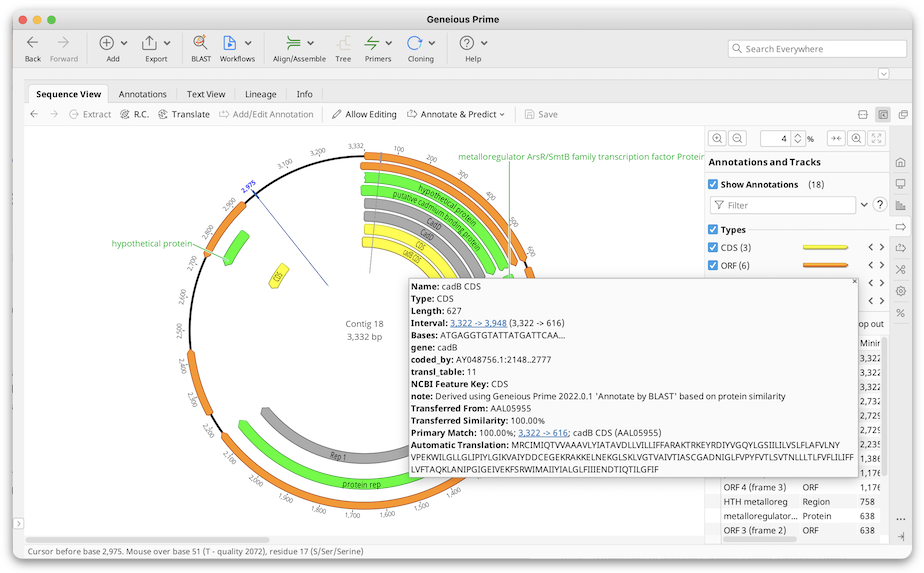
Transferred annotations will contain the annotation qualifiers from the original nucleotide sequence plus qualifiers detailing the source of the transferred annotation and the similarity percentage.
Note : If you have a large number of annotations, we suggest using custom BLAST instead of BLAST to NCBI as this can be extremely slow. Be aware that the larger the BLAST database, the slower the search will be.
See the following post for information on setting up custom BLAST: How can I BLAST against my own sequences or a database that isn’t part of NCBI?
Note that if you are using a custom BLAST database, it must be created from annotated sequences or preformatted database files from NCBI. Unannotated sequences such as those in a fasta file cannot be used for BLAST databases with this feature.
More options :
Under More options you can set the BLAST parameters such as E-value, word size, substitution matrix, and number of CPUs to use when running Custom BLAST.