The Geneious de novo assembler can be used to align pairs of forward and reverse Sanger sequences to create a single consensus sequence. This can be done in bulk using the sequence name to guide the assembly.
First, trim the poor quality sequence off your reads using Annotate and Predict - Trim Ends. Set the Error probability limit to 0.05 to trim bases with a quality score of less than approximately 13. Annotate the trimmed regions so that you can see what has been trimmed and adjust it if you wish.
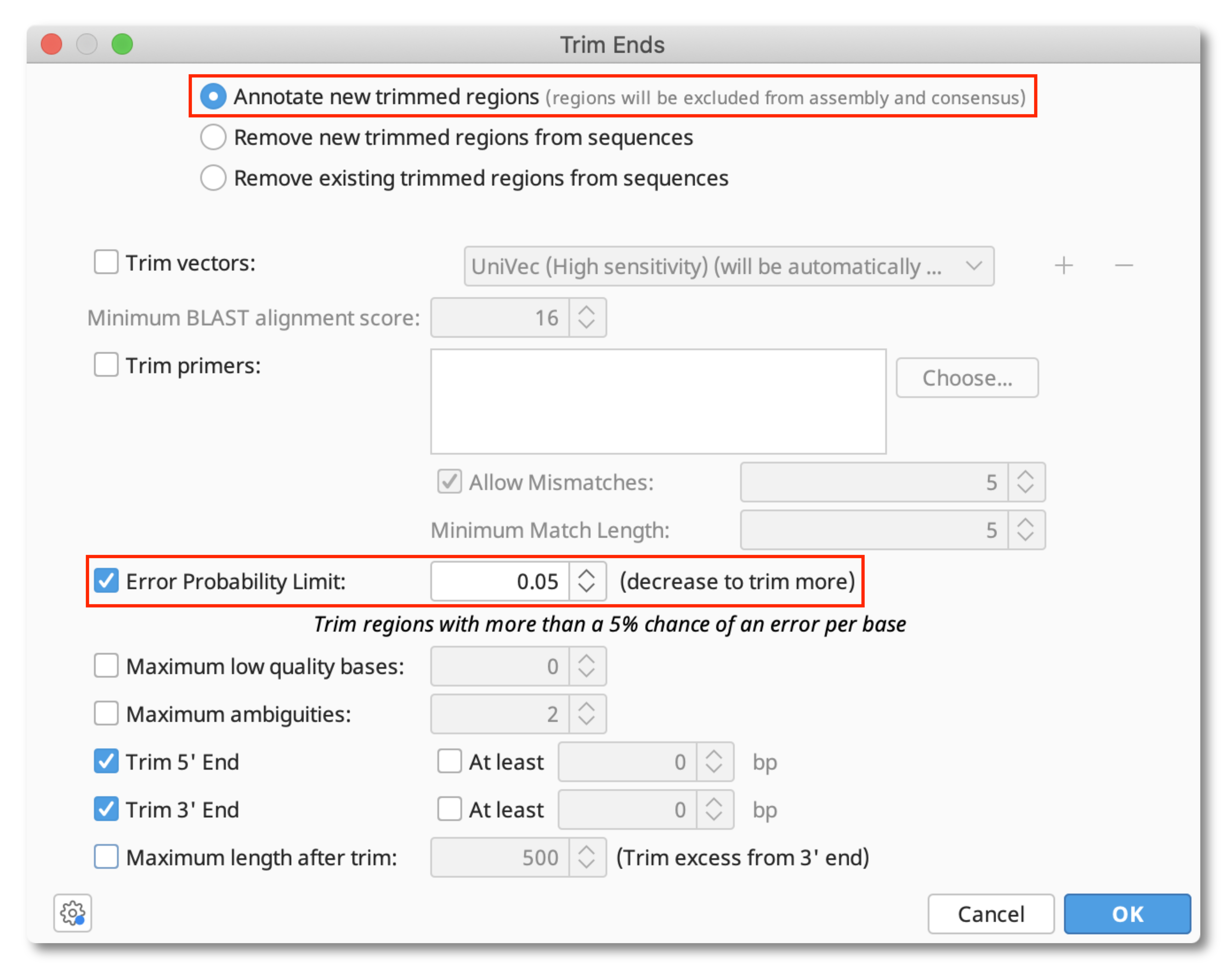
Save your changes after trimming.
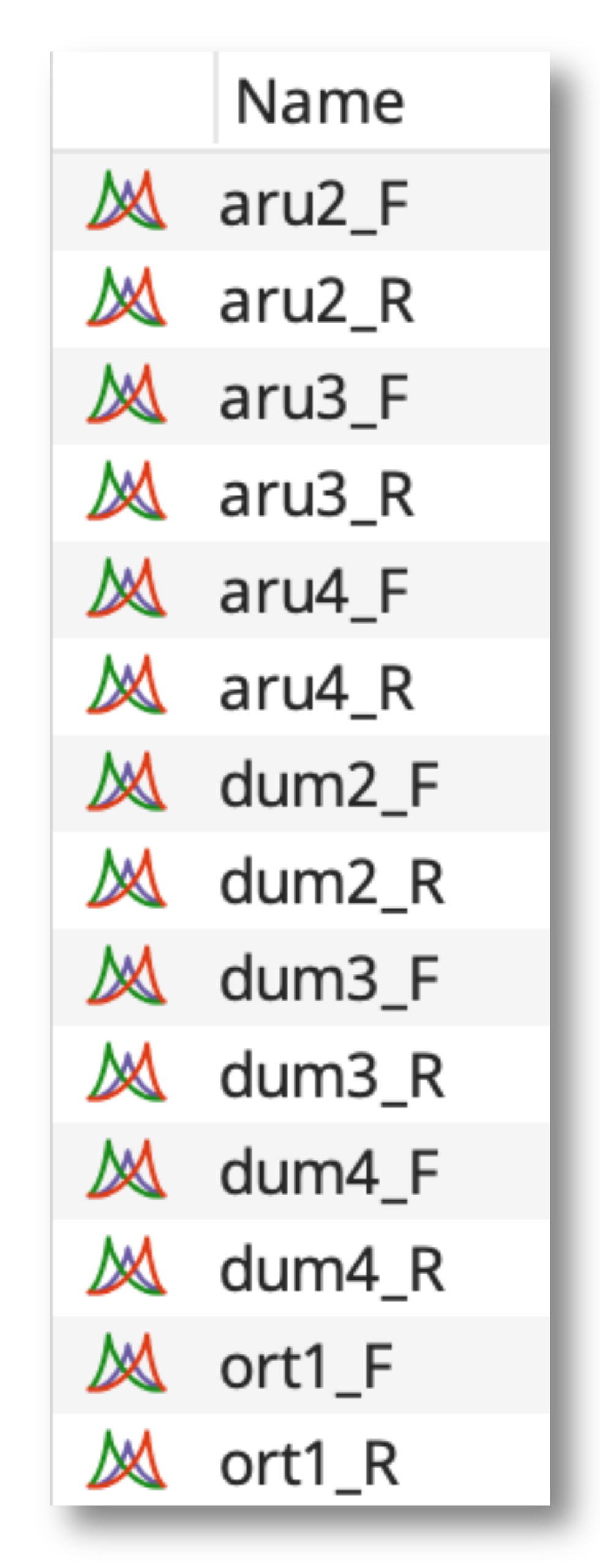
Next, go to Align/Assemble - De novo assemble to open the de novo assembly options. Select the option Assemble by name and then choose the part of your sequence name that is identical between the F and R pairs, plus the symbol that separates this part from the non-identical part of the name.
For example, for this list of F and R pairs, the identical part of the name (aru2, aru3 etc) is the first part, and the F and R identifier is separated by an underscore.
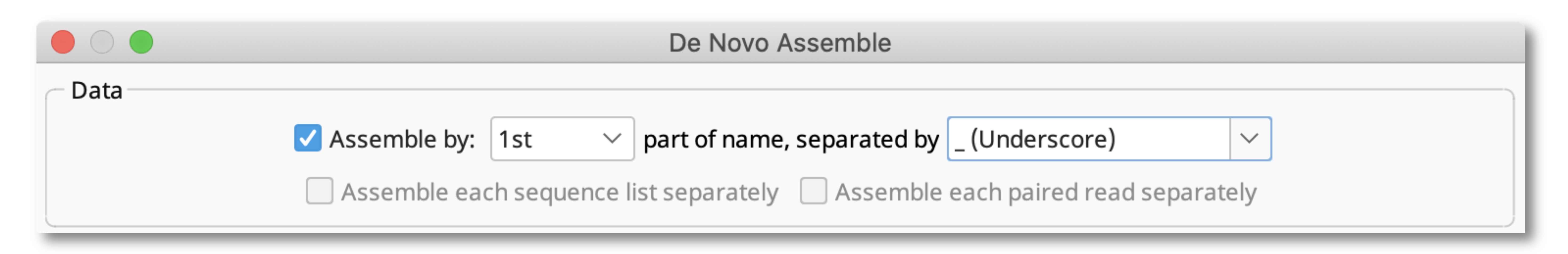
The de novo assembly settings should therefore be "Assemble by 1st part of name, separated by _ (Underscore)".
This will produce a contig document for each pair, as shown below.
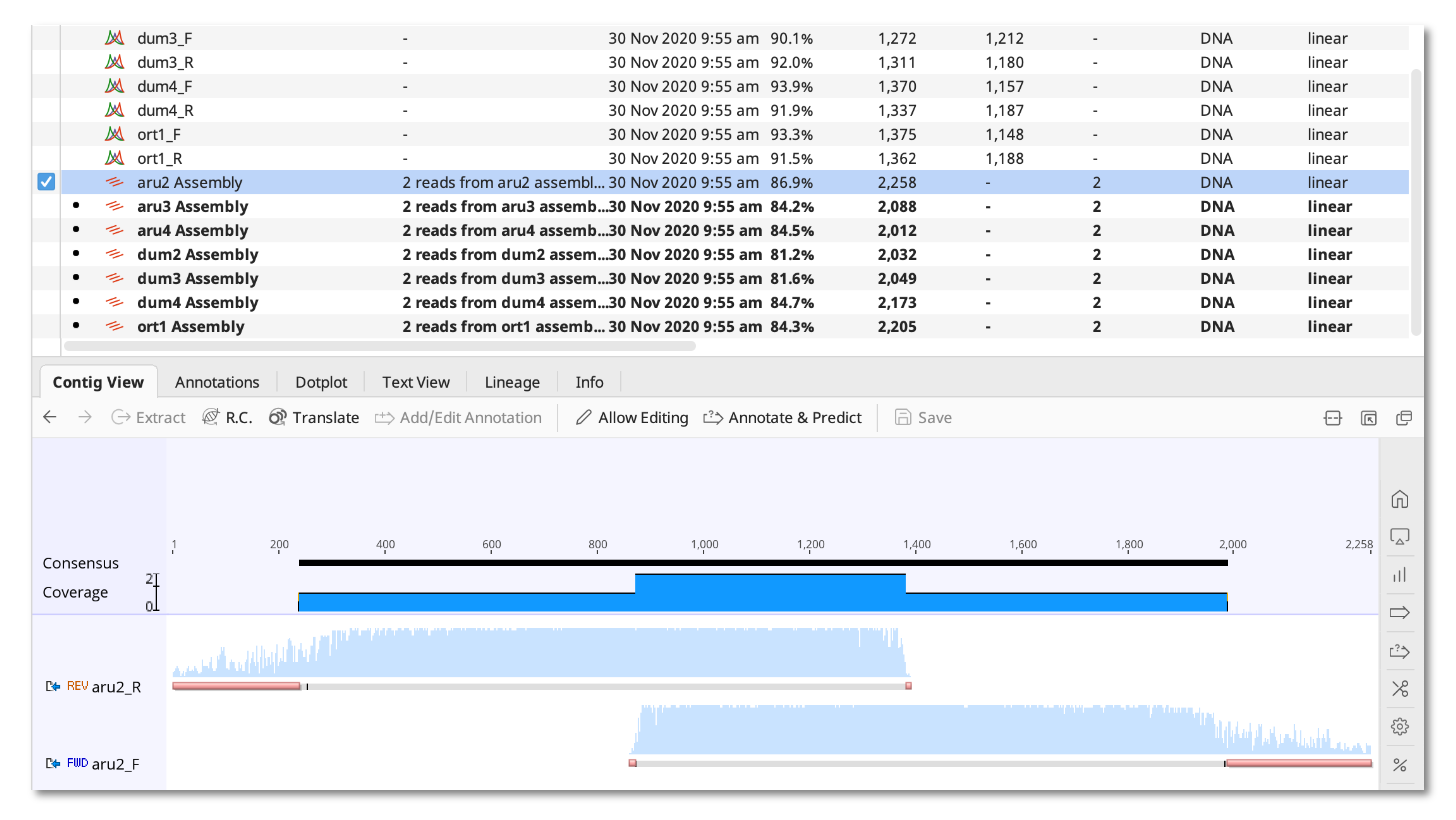
A consensus sequence for each pair of reads can be generated either during the de novo assembly process, by selecting the Save consensus sequences option in the de novo assembly setup window, or after the contigs have been generated and visually inspected. To generate a consensus sequence from the contig assembly documents, select all of the assemblies and go to Tools > Generate Consensus Sequences.