The Geneious Prime Variant caller, accessed via Geneious Prime menu Annotate & Predict - Find Variations/SNPs..., is primarily designed to call statistically validated variant positions based on an assembly of reads against a reference. This is because multiple sequences assembled against a reference are all expected to be "reads" derived from the same genome, and so contribute the same SNP information.
The Variant caller can be used to identify variant positions for an alignment of a consensus sequence and a reference sequence. However, in this case you should always align or assemble only two sequences, the reference and a consensus sequence, then call SNPs on the pairwise alignment/assembly.
Map to reference assembly
Provided your reference sequence and your consensus sequence are closely related and contain no significant insertions/deletions or rearrangements then you may be able to align them using the Geneious Map to Reference assembler. Select your reference and a single consensus "read" sequence and run Align/Assemble - Map to reference. If successful, this will output an assembly file. Be sure to specify the Geneious Mapper in the Map to reference setup, most other mappers available in Geneious Prime will not handle large input "read" sequences.
Pairwise alignment
As for map to reference assembly, provided your reference sequence and your consensus sequence are closely related and contain no significant insertions/deletions or rearrangements then you may also be able to align them with a pairwise aligner. For long sequences consider installing and using the MAFFT aligner (available as a plugin) as this is the fastest aligner available in Geneious Prime.
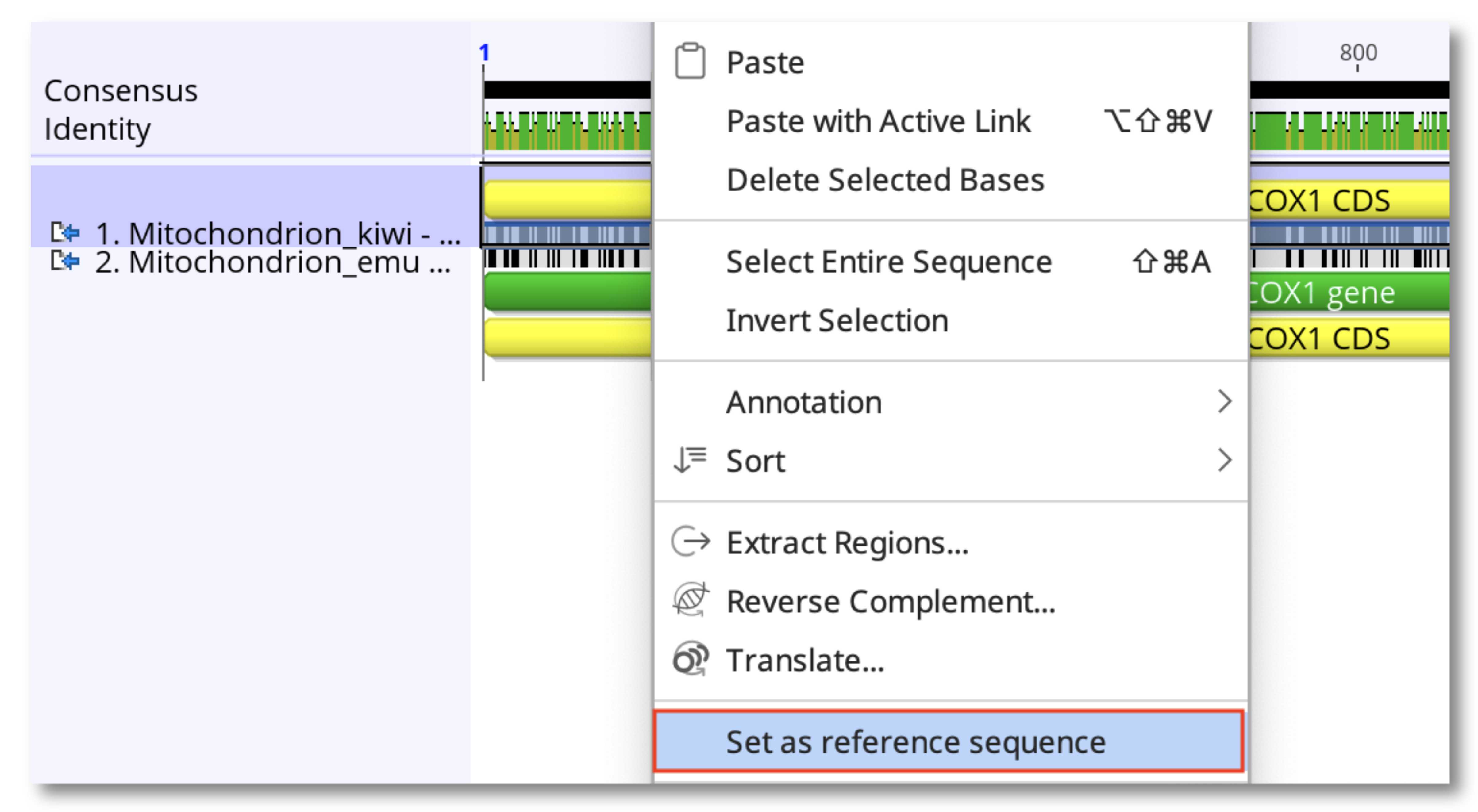
If you have created an alignment of two sequences with a pairwise aligner, then prior to calling variants/SNPs you will need to define the reference. To do this, select the alignment file, right-click on the name of the reference sequence in the alignment and choose Set as Reference sequence then save.
Identifying variant/SNP positions
Once you have created reference/single consensus sequence alignments or assembly files, you can then select each alignment and go menu Annotate & Predict - Find Variations/SNPs....
Make sure to uncheck options related to statistical validation as shown below as these options only apply to assemblies of raw sequence reads.
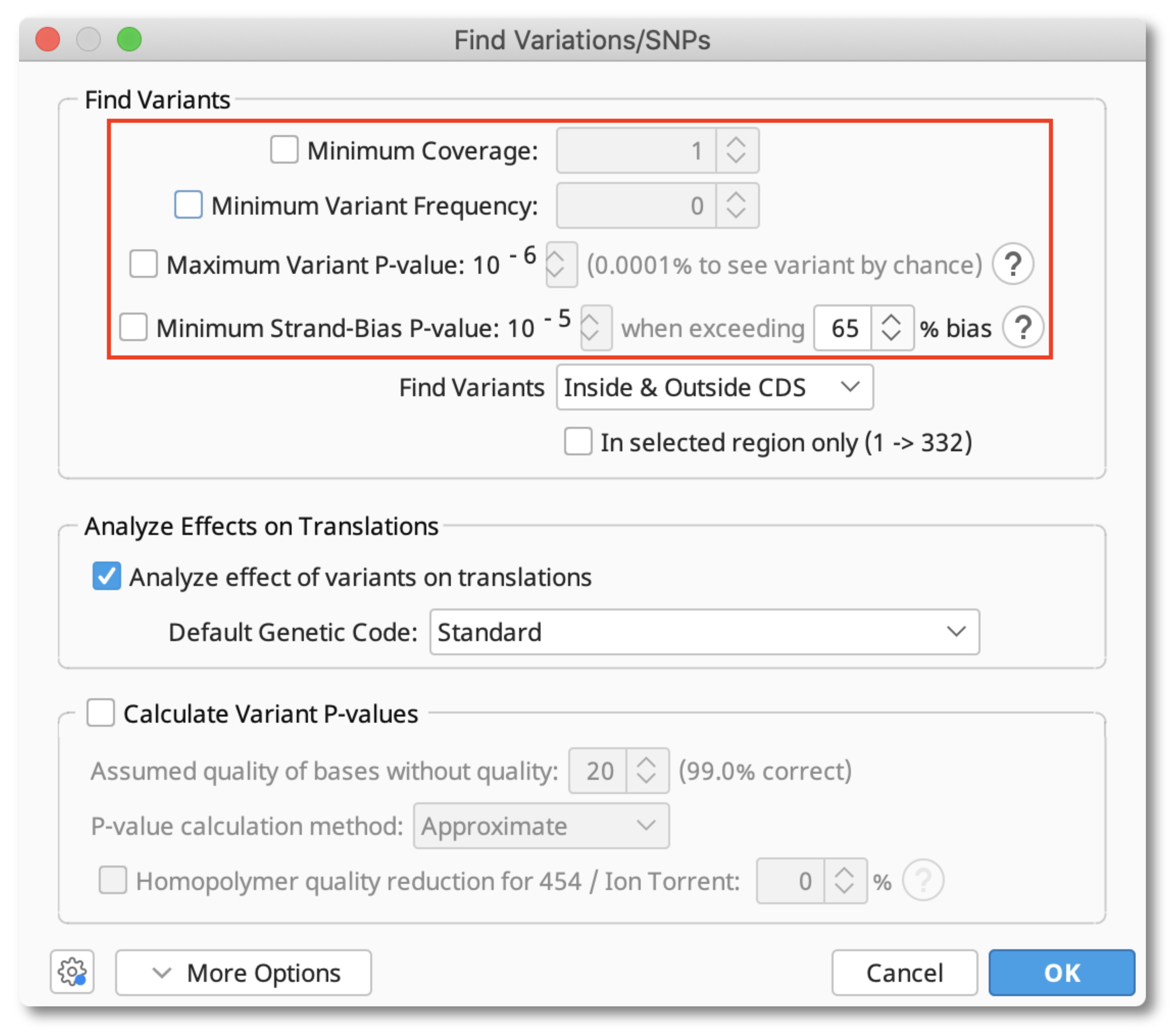
Any SNPs identified will be added to a track associated with the reference sequence. When you save ensure you choose to Apply changes to originals. The SNP track will then be added to the parent reference sequence.
If you have multiple consensus sequences, then align each separately with the reference, and call SNPs for each sequence assembly. Each time you save a new SNP track will be added back to the parent reference sequence.
Analysing large numbers of sequences
You can automate the above process using a Workflow. The attached "SNPs per sample" workflow (works with R11.0.3 or later) takes a reference and a selection of sequences (or a list of sequences) as input, performs pairwise assemblies of each sequence to the reference (saving them to a subfolder), then calls and adds SNP tracks to the assemblies and the linked reference sequence.
To use the Workflow, download then drag and drop the attached Workflow file into Geneious to import it. Then select your reference, and input sequences and go Toolbar - Workflows - SNPs per sample, define which input sequence is the reference then hit OK to start the assembly and SNP analysis.
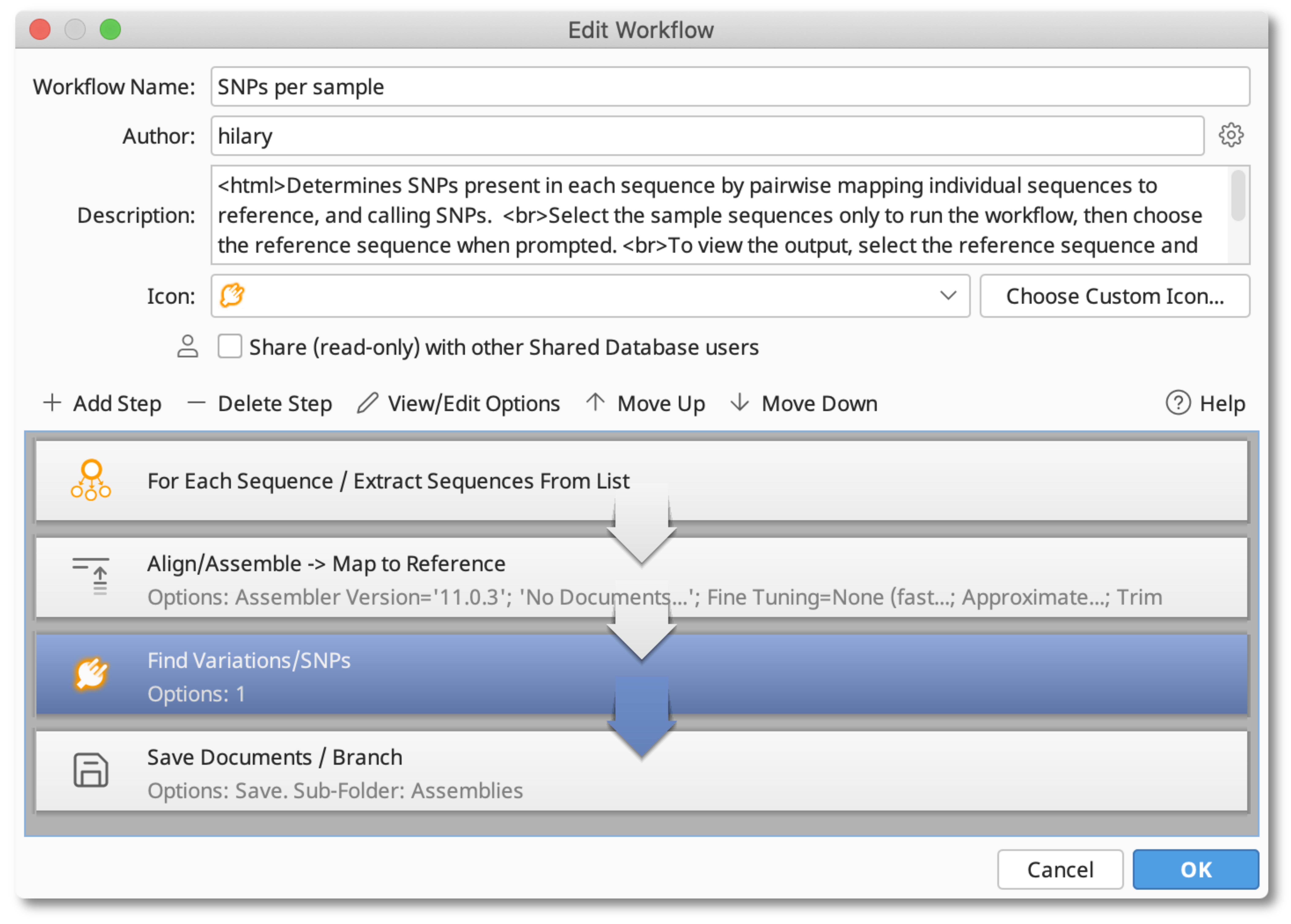
Once the Workflow has completed, select the reference sequence to view the New SNP tracks.
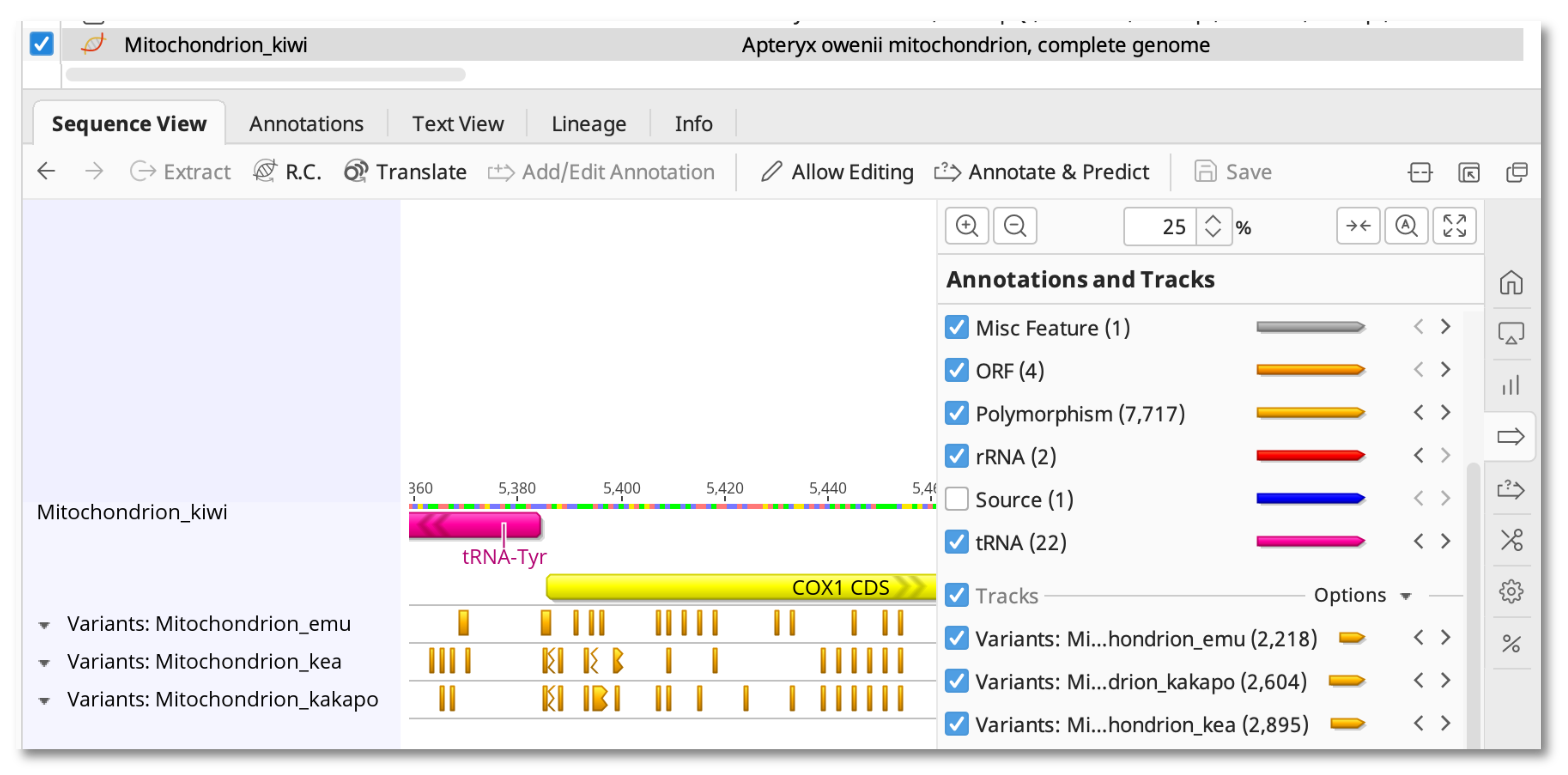
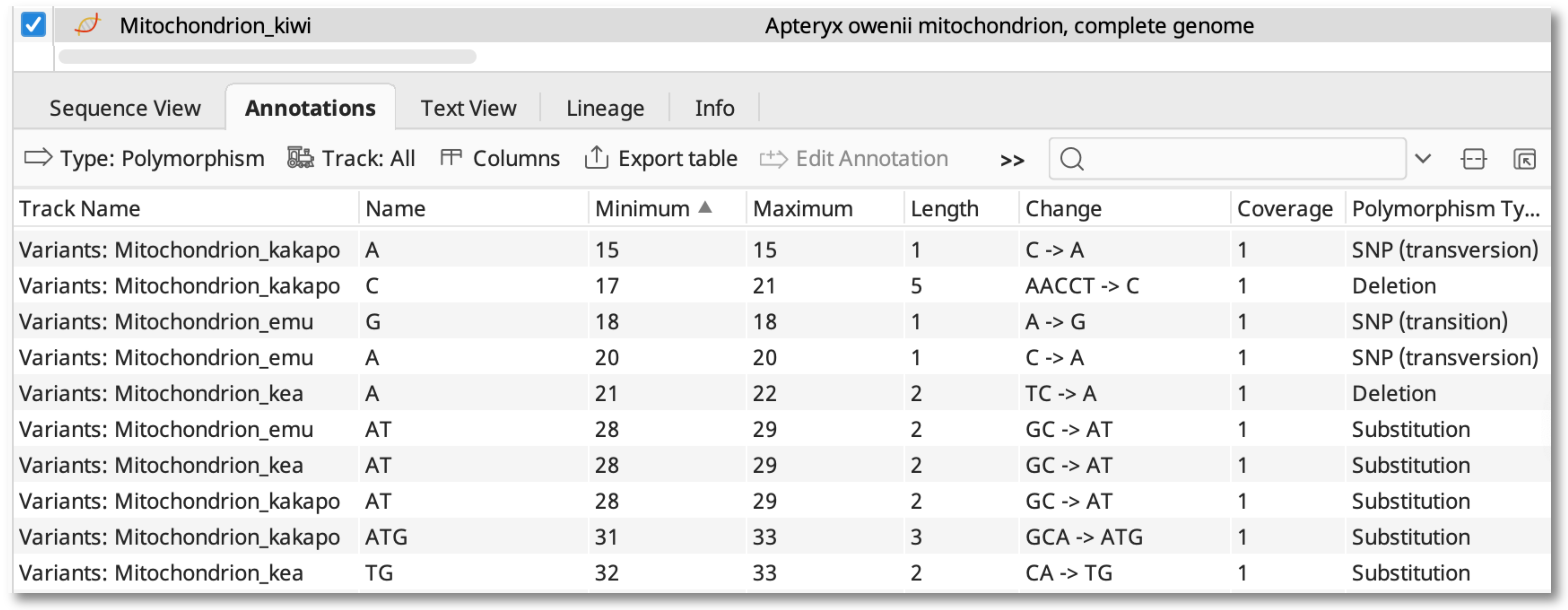
To view details of the SNP calls, switch to the Annotations table. Select Type: polymorphism, Track: All, and use the Columns button to display the column for Track Name.