It is possible to perform hybrid assemblies in Geneious using data from different sequencing technologies using the SPAdes de novo assembler. When datasets from long read (Nanopore or PacBio) or short read (Illumina) sequencing technologies are selected together as input, SPAdes will automatically run in hybridSPAdes mode.
In order for this to work, you will need to firstly specify the read type for each of your datasets. This either be done when the file is imported, or can be set afterwards using Sequence ->Select Read Technology...
Once the read technology is set, you can see it displayed in the document viewer under the Read Technology heading, as shown below.

You can then run the de novo assembly by selecting your multiple documents and going to the Align/Assemble>De Novo Assemble menu. Select SPAdes as your assembler and this will bring up the following menu:
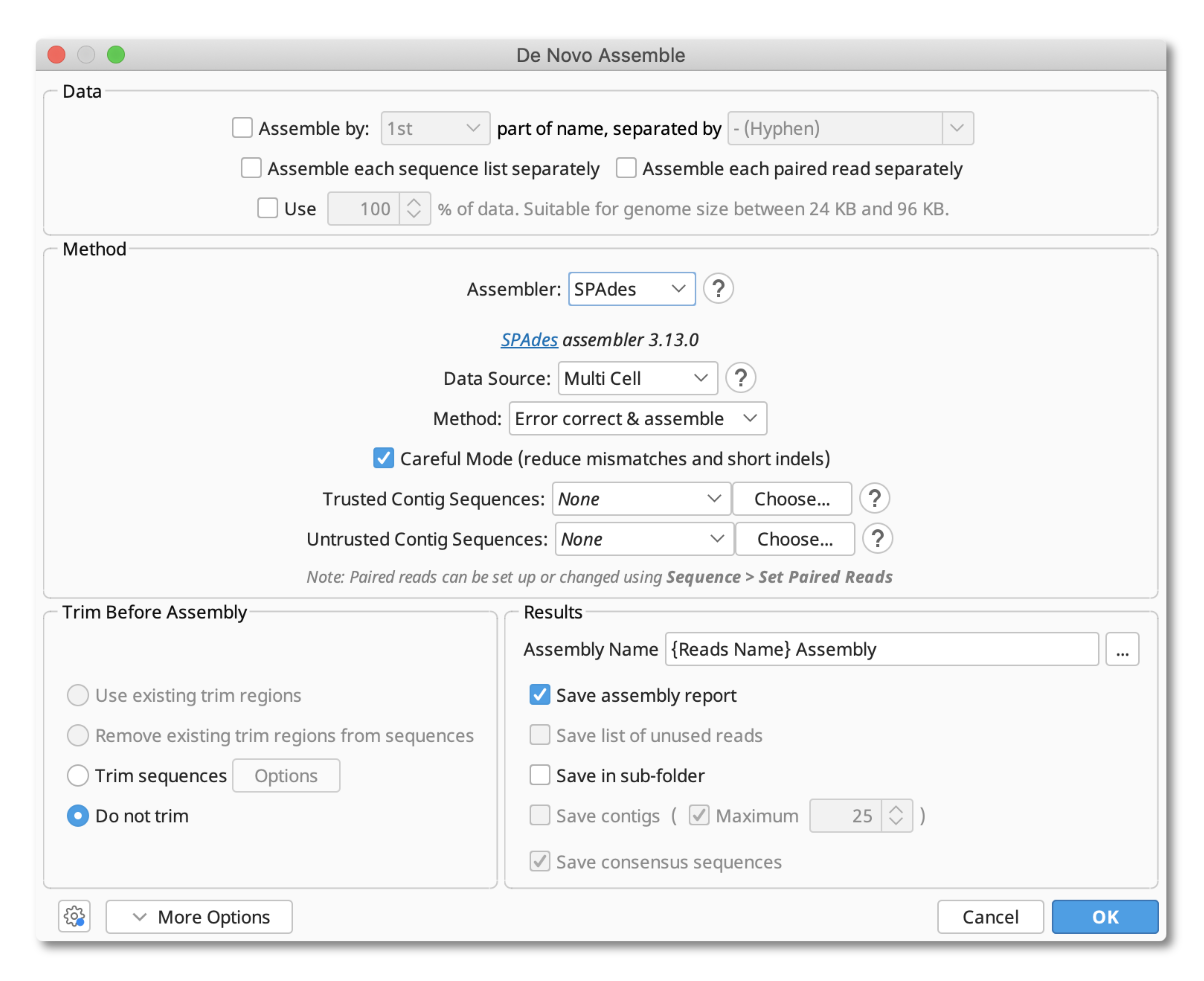
For hybrid assemblies, the data source should be set to Multi Cell or Single Cell (Metagenome, RNA and Plasmid settings are not supported for hybrid datasets).
Under Method it is possible to choose whether or not you wish for an Error correction step to be included in your assembly. Note that error correction is only for Illumina reads.
Specifying careful mode will try to reduce the number of mismatches and indels in your assembly. This will also run MismatchCorrector, a post-processing tool.
Optional: If you have a set of contigs from another assembly you wish to include in your dataset, you can add these as Trusted or Untrusted contigs. Only contigs from the same genome should be specified. Trusted contigs are high quality contigs, while untrusted contigs may have more errors or be of unknown quality.
Change the settings in the de novo assembly menu as appropriate for your data set, then select OK to run the assembly.